Overview
This R&D Briefing presents the Centre for Innovation in Regulatory Science (CIRS) annual analysis of new active substance (NAS) approvals by six major regulatory authorities: the European Medicines Agency (EMA), US Food and Drug Administration (FDA), Japan Pharmaceuticals and Medical Devices Agency (PMDA), Health Canada, Swissmedic, and the Australian Therapeutic Goods Administration (TGA). The analysis focuses on approvals granted in 2025 while examining trends observed over the last decade (2016–2025).
The briefing explores trends in regulatory approval timelines, submission gaps, expedited review pathways, facilitated regulatory pathways, orphan designations, therapeutic areas, common approvals across agencies, FDA use of patient experience data, and the individual components of EMA regulatory timelines.
Why This Matters
Regulatory approval timelines are often used as an indicator of how quickly new medicines become available to patients. However, comparisons between regulatory authorities can be influenced by a range of factors beyond the scientific review itself. These include company submission strategies, the timing of submissions across markets, product characteristics, and the use of expedited review pathways and facilitated regulatory pathways.
This annual CIRS analysis examines trends in new active substance approvals over a ten-year period (2016–2025), providing a broader perspective on evolving regulatory practices and the factors that may influence the availability of innovative medicines globally. The briefing also explores how expedited review pathways, orphan designations, and facilitated regulatory pathways are shaping the regulatory landscape across major authorities.
Key Findings
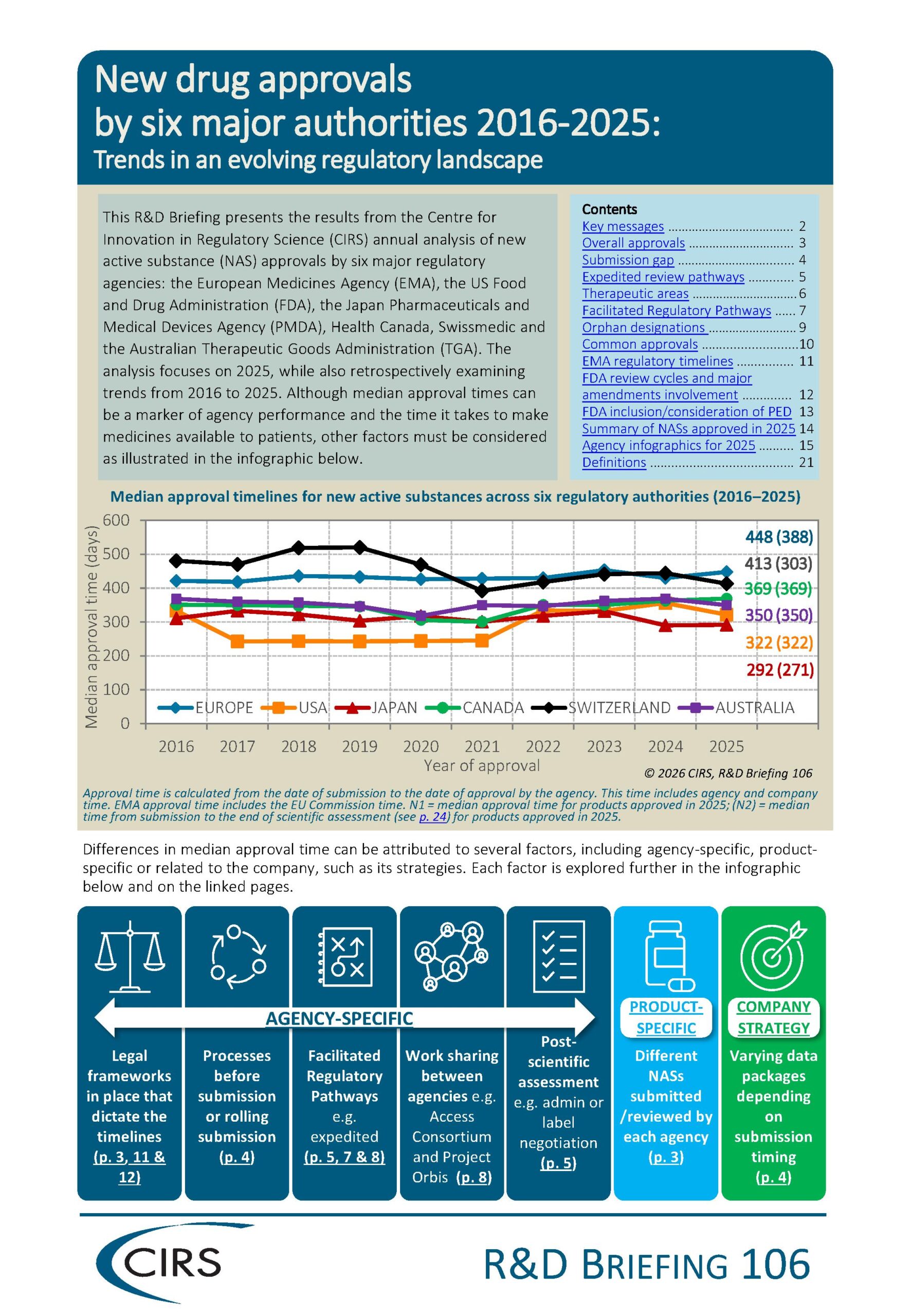
- In 2025, FDA granted the highest number of new active substance approvals (50), followed by Health Canada (44), TGA (42), PMDA (40), EMA (39), and Swissmedic (34).
- PMDA recorded the shortest median approval time in 2025 (292 days), followed by FDA (322 days), TGA (350 days), Health Canada (369 days), Swissmedic (413 days) and EMA (448 days).
- Expedited review pathways were associated with shorter approval times than standard reviews across all six regulatory authorities.
- The use of facilitated regulatory pathways increased across all six authorities between 2021–2025 compared with 2016–2020.
- The number of new active substances approved by all six authorities increased from 37 in 2016–2020 to 52 in 2021–2025, reflecting continued growth in global approvals.
What You’ll Find in This Briefing
This publication includes:
- Approval timelines and submission gap trends across EMA, FDA, PMDA, Health Canada, Swissmedic, and TGA.
- Analysis of expedited review pathways and their impact on regulatory approval times.
- Trends in facilitated regulatory pathways, including collaborative review and work-sharing initiatives.
- Comparisons of approval timelines by therapeutic area and orphan designation status.
- Assessment of medicines approved by all six regulatory authorities and evolving global submission strategies.
- Insights into FDA review cycles and the inclusion of patient experience data in regulatory decision making.
- Detailed breakdowns of EMA regulatory timelines, including scientific assessment, company response, validation, and European Commission decision times.
Access the Resources
- Download R&D Briefing 106
- Download the 2025 NAS List
Questions?
If you have any questions or comments on this study, please get in touch with Dr Magda Bujar: mbujar@cirsci.org